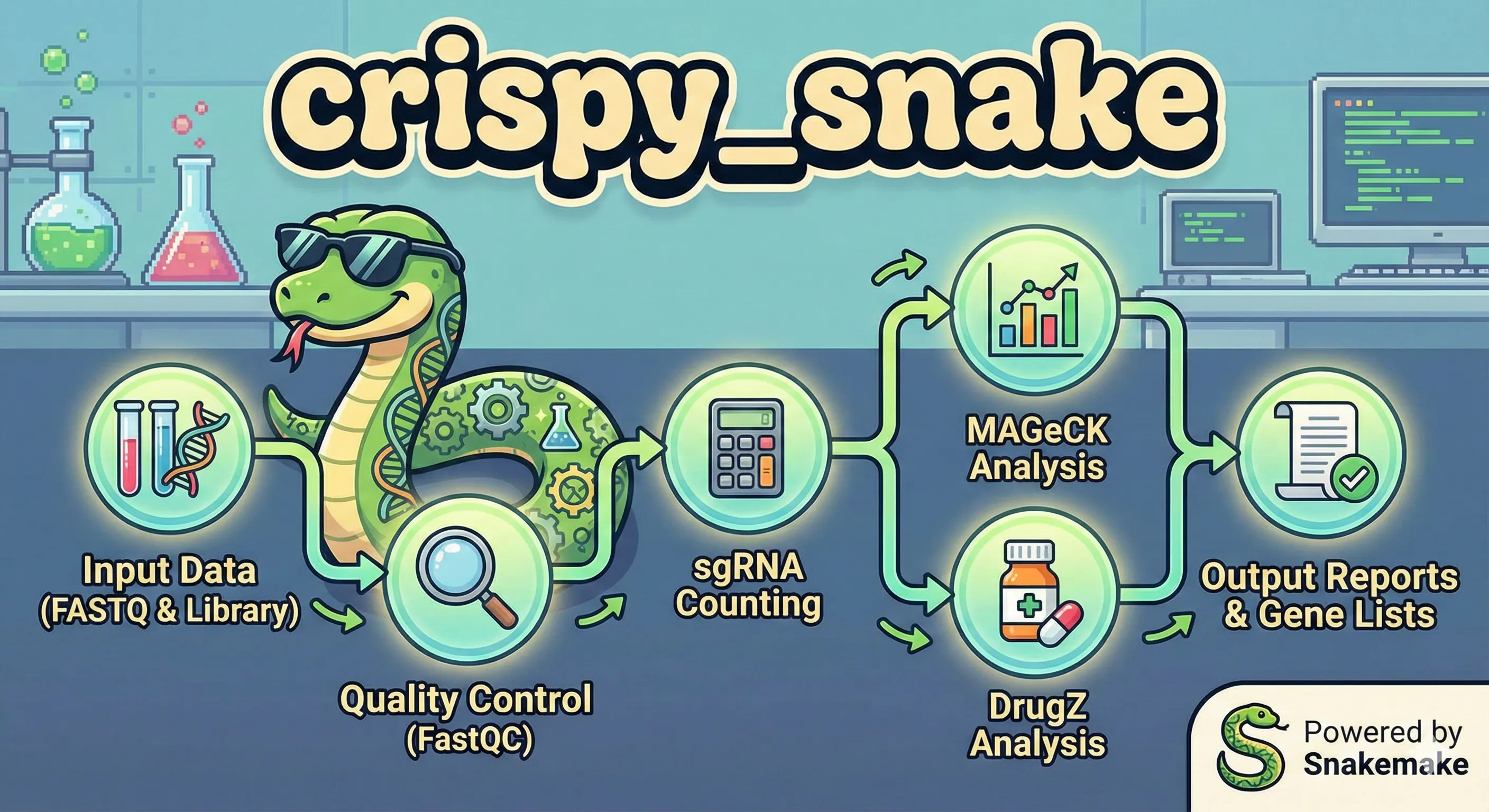
Project #4: crispy_snake - CRISPR Screen Analysis with Snakemake
GitHub Repo
Built With: Snakemake | Python 3 | FastQC | MAGeCK | DrugZ | Conda
Practical Objective: Automate the processing and analysis of CRISPR knockout screens, taking raw FASTQ sequencing data to statistical hit identification and visualization.
Learning Objective: Build fluency with workflow management systems to create accessible and scalable bioinformatics pipelines.
Purpose of crispy_snake
CRISPR screens generate large datasets that require multi-step processing: quality control, read trimming, alignment, counting, and statistical modeling. Running these steps manually via shell scripts is error-prone and intimidating to bench scientists.
I built crispy_snake for my lab to enable in-house processing and analysis of CRISPR screens with a human-readable config file and single command. Snakemake scales easily across environments, making it perfect for use on the Yale HPC clusters.
Workflow Architecture
The pipeline is built on Snakemake, which defines the rules of the analysis as a Directed Acyclic Graph (DAG). This offers several advantages over bash scripts:
- Dependency Management: Each step runs in an isolated Conda environment. This prevents version conflicts (Python 2.7 for an old tool vs. Python 3.10 for a new one).
- Parallelization: Snakemake automatically parallelizes independent jobs (like processing four replicates at once) based on available CPU cores.
- Input Validation: The pipeline enforces compatibility at startup. This prevents silent failures common in legacy bioinformatics tools.
Dual Statistical Modeling
Due to inherent variability of biological data and the many-hypotheses nature of screening approaches, custom statistical methods (MAGeCK, DrugZ) are utilized for CRISPR screens. crispy_snake runs both algorithms in parallel:
- MAGeCK: Uses a negative binomial model to identify essential genes. It is highly robust to noise/outliers because it relies on consistent guide rankings rather than raw magnitude.
- DrugZ: Uses a normalized Z-score approach. It relies on the summed magnitude of fold-change, typically increasing sensitive for detecting gene depletion.
Modernizing DrugZ
The original DrugZ algorithm is written in Python 2 and unavailable on standard package managers, so I refactored the codebase for Python 3/Pandas 2.0.
Crucially, I added native support for non-targeting control (NTC) centering. While whole-genome screens assume the median guide has zero biological effect, targeted libraries often violate this assumption. For example, a DNA damage response (DDR) screen will have a median shifted toward lethality (negative LFC). A tumor-suppressor screen will have a median shifted toward enhanced proliferation (positive LFC).
crispy_snake corrects this bias by centering the distribution on the NTCs, recovering hits that would otherwise be masked by the shifted distribution.
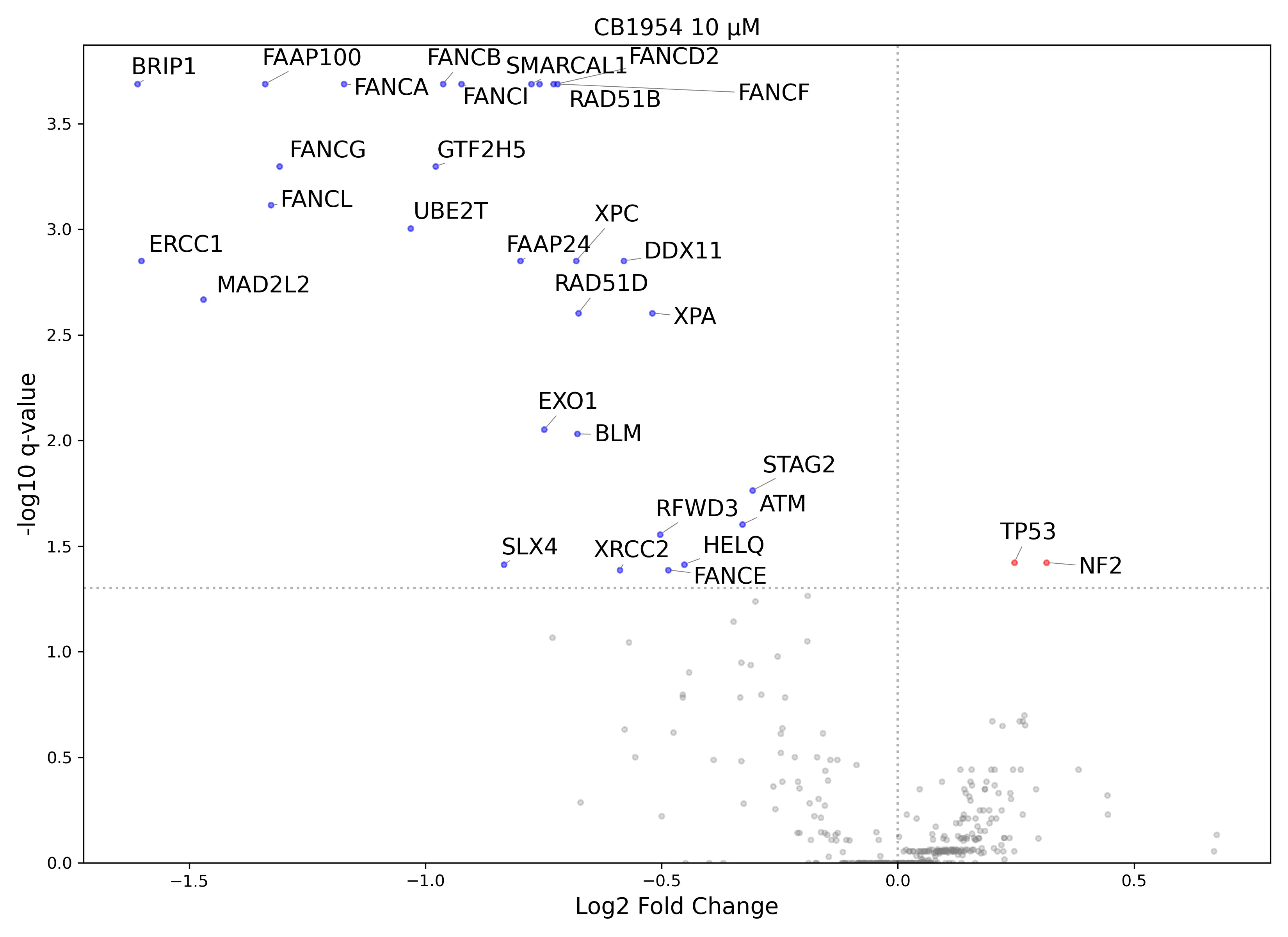
Automated Visualization
In addition to data tables and quality reports, the pipeline automatically generates volcano plots for each comparison defined in the config file (e.g. vehicle vs. treated). It highlights significant hits (FDR < 0.05) in red (enrichment) or blue (depletion), allowing for immediate visual assessment of the screen's results.